Written by: Jackson Udy, PE
Process Engineer at Process Engineering Associates
May 12, 2025
One common decision made during the front-end engineering and design (FEED) of a project is selecting the appropriate means of heat transfer to and from the process. However, selecting a heat transfer fluid can feel difficult due to the many options available. This article outlines the benefits and drawbacks of the most common heat transfer fluids and quantitatively compares various heat transfer fluids for heating, cooling, and refrigeration service.
Introduction
Heat transfer fluids are used for a variety of services in the process industry such as:
- Fluid heating and vaporization
- Fluid cooling, condensing, and crystallization
- Reboiling liquids for distillation
- Combined Heating and Cooling of Batch Systems
- And many other additional services
The variety of process conditions encountered in the process industry means that the optimal heat transfer fluids may be different for each situation. Proper selection of heat transfer fluids results in lower capital and operating costs, and process engineers should evaluate the choice of heat transfer fluid carefully. The sections below describe the benefits and drawbacks of the most common fluids to help provide a guide for selecting a good heat transfer fluid.
Steam
Steam is an inexpensive and widely available heat transfer fluid. Due to its high film coefficient, using steam often results in smaller heat exchanger sizes. Steam is also available at most process plants and, if additional boiler capacity is available, it can be a very economical option.
Steam is quite useful for heating fluids to temperatures as high as 450°F. However, as process temperature requirements increase past 450°F, the applicability of steam becomes problematic. The vapor pressure of steam increases exponentially, and very high pressure is required to meet process temperatures above 450oF. For example, saturated steam at 600°F has a vapor pressure of ~1,530 psig. A typical hot oil system operating at the same temperature will often operate below 150 psig. Thus, using steam for high temperature applications often requires more expensive equipment and piping compared to other available heat transfer fluid options.
Another item that should be considered with steam use is when steam is being used in a dual-use jacket or coil, which are commonly applied to batch reactors. Dual-use jackets use steam for heating and then switch to cooling water service for cooling. Swapping between steam and cooling water, for example, can lead to hammering in equipment jackets and coils due to the rapid condensation of steam. This can cause additional stress on equipment and should be considered if using steam in a system requiring both heating and cooling. Some designers will provide separate heating and cooling coils for batch systems, but this comes at an additional capital cost.
Finally, steam systems typically require chemical treatment to limit corrosion and condensate recovery systems to route condensate back to the boiler. These systems can add significant maintenance costs to the steam system.
Water
Water is the most common fluid used for process cooling in the process industries. Water is cheap and widely available. Additionally, water can be cooled by evaporative cooling via cooling towers, eliminating the need for refrigeration units. Some locations may even allow once through water cooling, where water is pulled directly from nearby bodies of water for process cooling and then returned to the body of water.
Water’s advantages are its low cost and high heat transfer coefficient. Water is orders of magnitude cheaper than other commercial heat transfer fluids. Additionally, water almost always has a higher heat transfer coefficient than heat transfer oils, glycol/water mixtures, or brine systems.
One of the main disadvantages of water is its limited operating range. Cooling water systems operating above 120°F are often subject to increased scaling and fouling. Typical supply temperatures for cooling water systems are about 85°F depending on the season and location, and chilled water systems cannot operate below 32°F. Water can also be highly corrosive and promote biological growth, therefore carefully monitored chemical treatment systems are often required to mitigate these issues. Finaly, cooling water systems often require substantial water blowdown rates that may require discharge permitting and have other environmental considerations.
Heat Transfer Oils
Organic heat transfer fluids systems are common in many industries. The main advantage of organic heat transfer fluids in heating services is the reduced operating pressure compared to steam and subsequent reduced equipment costs. Many organic heat transfer fluids can also be used in cooling service, providing flexibility for batch processes. Heat transfer oils require no blowdown and may operate for many years without being changed out if operated within their recommended temperature range. Some heat transfer oils are designed to operate in chilled service, well below the freezing point of water. Most modern heat transfer oil systems operate in the liquid phase, but vapor phase systems are not uncommon. Due to the low vapor pressure and good stability of many heat transfer oils, many systems are operated open to the atmosphere and use equipment with low pressure ratings. The main drawback of heat transfer oils is their reduced heat transfer coefficient compared to steam and cooling water systems. Another drawback is the cost of heat transfer oils compared to water and glycol-based fluids. Most heat transfer oils are also combustible, which may be a drawback for some facilities.
Brine and Glycol Systems
Calcium chloride (CaCl2) brine systems are water-based heat transfer fluids that decrease the freezing point of water and allow for use in low temperature applications.
CaCl2 brine has a higher heat transfer coefficient but can be more corrosive to piping than glycol systems and requires chemical inhibitor programs to ensure mechanical reliability. However, with the right inhibitors and proper monitoring in place, carbon steel can be used in calcium chloride brine systems. Glycols on the other hand have much lower corrosion rates than salt brines, particularly inhibited glycols. Ethylene Glycol water mixtures are generally the preferred glycol due to low viscosity and increased heat transfer rates. Propylene glycols have lower heat transfer coefficients but are generally considered not toxic and are often used in food and beverage industry where incidental food contact is possible. Glycols also have higher maximum operating temperatures than brines, giving them some extra flexibility as they can be used for both cooling and low temperature heating below 300°F.
Selecting a Heat Transfer Fluid
Selecting a heat transfer fluid involves balancing a variety of concerns. Some of the factors to consider when selecting a heat transfer fluid are:
- Service temperature
- Film coefficient
- Fluid Cost
- Fluid Vapor Pressure
- Waste generation, blowdown requirements
- Food Contact Rating Required?
A primary concern when selecting a heat transfer fluid is to ensure a good heat transfer film coefficient at the required service temperature. Tables 1, 2, and 3 below show the heat transfer coefficients for various heat transfer fluids flowing at 6 ft/s inside ¾” tubes at 70°F, 20°F, and 400°F respectively. Various heat transfer fluids are shown in Tables 1, 2, and 3 to give a quantitative example of the tradeoffs between different fluids. Note that viscosity is usually the best predictor of heat transfer coefficient and, when looking for a quick evaluation between fluids, looking at viscosity will usually give a general idea of relative heat transfer coefficients.
Table 1: Heat Transfer (Film) Coefficients for Cooling at 70°F, 6 ft/s in 3/4″ ST HX tubes
| Property | Heat Transfer Fluid | |||||
| Cooling Tower Water | 50% Ethylene Glycol | 25% CaCl2 Brine | Dowtherm J | Paratherm MR | Therminol 66 | |
| Viscosity | 1.0 | 3.9 | 2.4 | 0.9 | 12.0 | 101.0 |
| Reynolds Number | 34750 | 9570 | 17950 | 32800 | 2330 | 350 |
| Heat Capacity | 1.00 | 0.78 | 0.70 | 0.44 | 0.60 | 0.38 |
| Thermal Conductivity | 0.35 | 0.22 | 0.33 | 0.07 | 0.07 | 0.07 |
| Heat Transfer Fluid Film Coefficient | 1329 | 189 | 880 | 320 | 44 | 32 |
Table 2: Heat Transfer (Film) Coefficients for Cooling at 20°F, 6 ft/s in 3/4″ ST HX tubes
| Property | Heat Transfer Fluid | |||
| 50% Ethylene Glycol | 25% CaCl2 Brine | Dowtherm J | Paratherm MR | |
| Viscosity | 10.9 | 6.1 | 1.4 | 29.0 |
| Reynolds Number | 3520 | 7160 | 22120 | 980 |
| Heat Capacity | 0.75 | 0.68 | 0.42 | 0.55 |
| Thermal Conductivity | 0.21 | 0.32 | 0.08 | 0.08 |
| Heat Transfer Fluid Film Coefficient | 139 | 230 | 288 | 37 |
Table 3: Heat Transfer (Film) Ceofficients for Heating at 400°F, 6 ft/s in 3/4″ ST HX Tubes
| Heat Transfer Fluid | ||||
| Property | Steam | Dowtherm J | Therminol 66 | Paratherm MR |
| Viscosity | 0.2 | 0.8 | 0.6 | |
| Reynolds Number | 89170 | 28620 | 47340 | |
| Heat Capacity | 0.59 | 0.53 | 0.66 | |
| Thermal Conductivity | 0.05 | 0.06 | 0.07 | |
| Heat Transfer Fluid Film Coefficient | 1500 | 432 | 280 | 440 |
| Vapor Pressure | 233 | 25.3 | 0.37 | 0.47 |
While film coefficients for each fluid can drive the economics of heat transfer fluid selection, some other factors to consider when selecting heat transfer fluids include:
- The required service temperature.
- -Does the fluid need to operate in both heating and cooling service?
- Fluid cost.
- -Generally synthetic heat transfer fluids are the most expensive, with water-based fluids being more economical.
- Process side film coefficient.
- -If the process side has a very low film coefficient (Such as vacuum condensing service or cooling a viscous liquid), then the process side may be the controlling resistance to heat transfer. In this scenario, selecting the heat transfer fluid with the highest film coefficient may not provide additional heat transfer, and choosing a more economical fluid with a lower film coefficient may make more sense.
- Is HT-1 food contact rating required?
- -Some processes require heat transfer fluids to be rated for incidental food contact in case of a leak.
- Does significant wastewater generation or discharge permits need to be avoided?
- – This may preclude steam or cooling water systems.
Final Thoughts
The variety of process conditions in industry means that the optimal heat transfer fluids are different for each process and location. Heat transfer fluids selection can have a significant effect on capital and operating costs and should be evaluated carefully. If you need a heat transfer system designed, or are having issues with an existing system, reach out to us at Process Engineering Associates. We have extensive experience with heat transfer fluids in a variety of industries. Give us a call to see how we can potentially help you select the best fluid for your application.


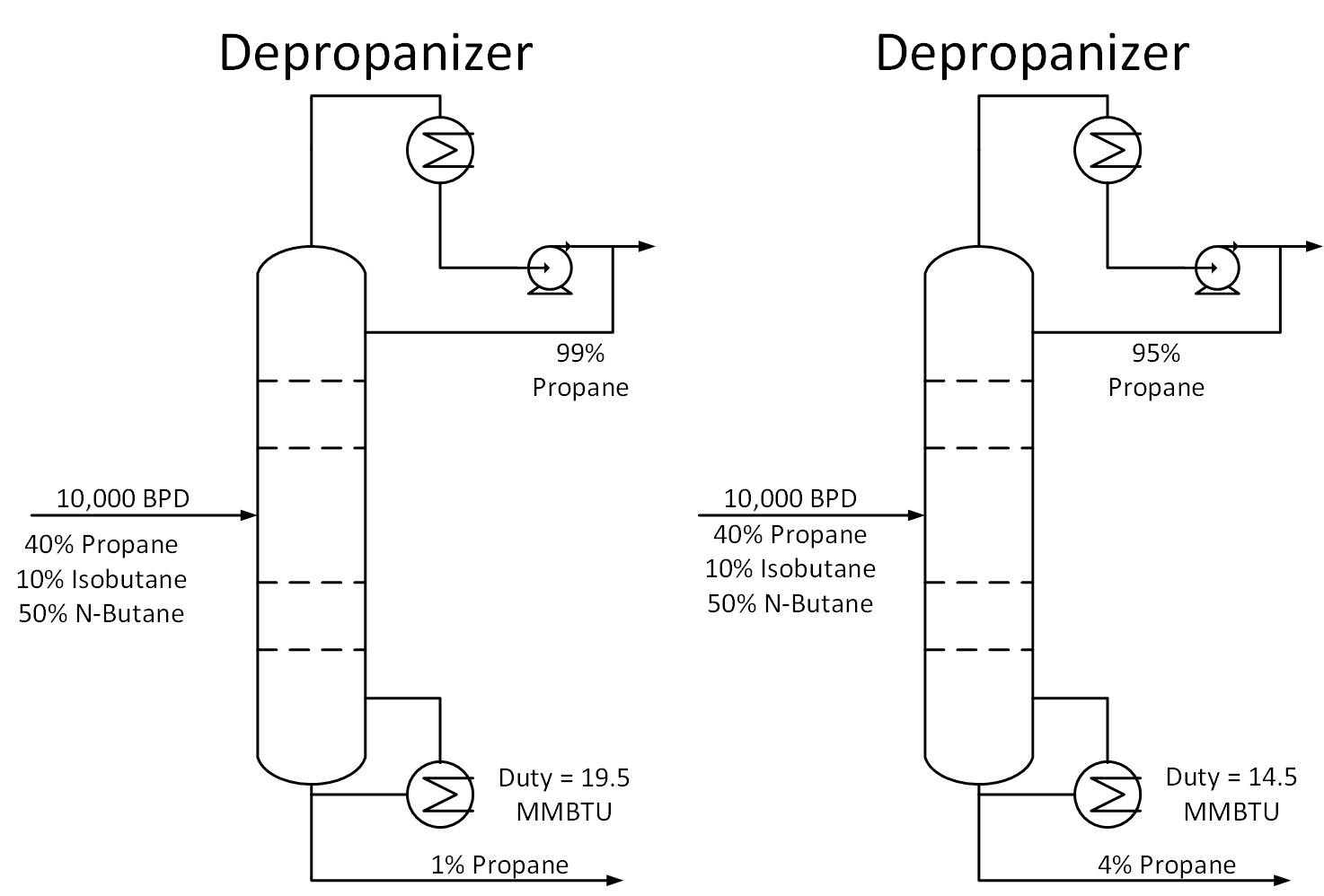 Typically operators will target above the required product purity to absorb any swings in tower operation while keeping product streams on spec. Reducing this ‘overshoot’ as much as feasible can result in great energy savings.
Typically operators will target above the required product purity to absorb any swings in tower operation while keeping product streams on spec. Reducing this ‘overshoot’ as much as feasible can result in great energy savings.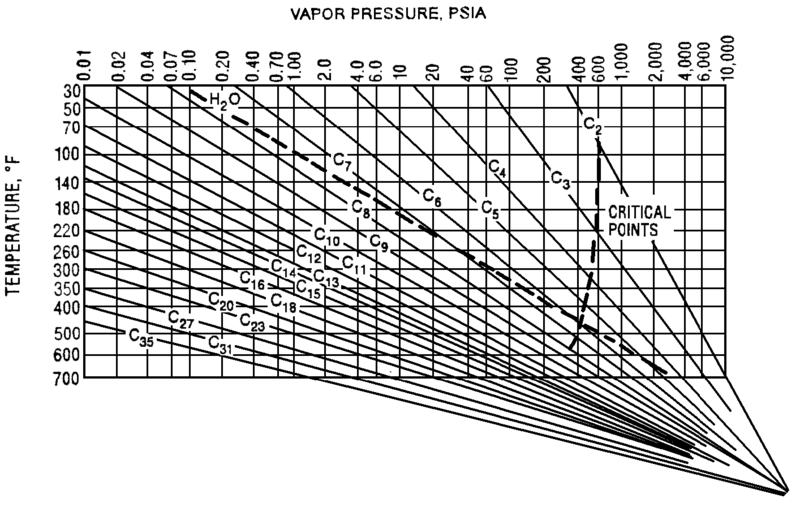 The Cox Diagram(3) above shows why decreasing column pressure reduces energy consumption. For any given compound, as column pressure decreases, the flash temperature also decreases. The diagram above shows the relationship between flash temperature and vapor pressure. As flash temperature decreases, so does the vapor pressure for each hydrocarbon. But more importantly, the difference in vapor pressure between the compounds increases. For example at 180 deg F, The relative volatility between propane and butane is
The Cox Diagram(3) above shows why decreasing column pressure reduces energy consumption. For any given compound, as column pressure decreases, the flash temperature also decreases. The diagram above shows the relationship between flash temperature and vapor pressure. As flash temperature decreases, so does the vapor pressure for each hydrocarbon. But more importantly, the difference in vapor pressure between the compounds increases. For example at 180 deg F, The relative volatility between propane and butane is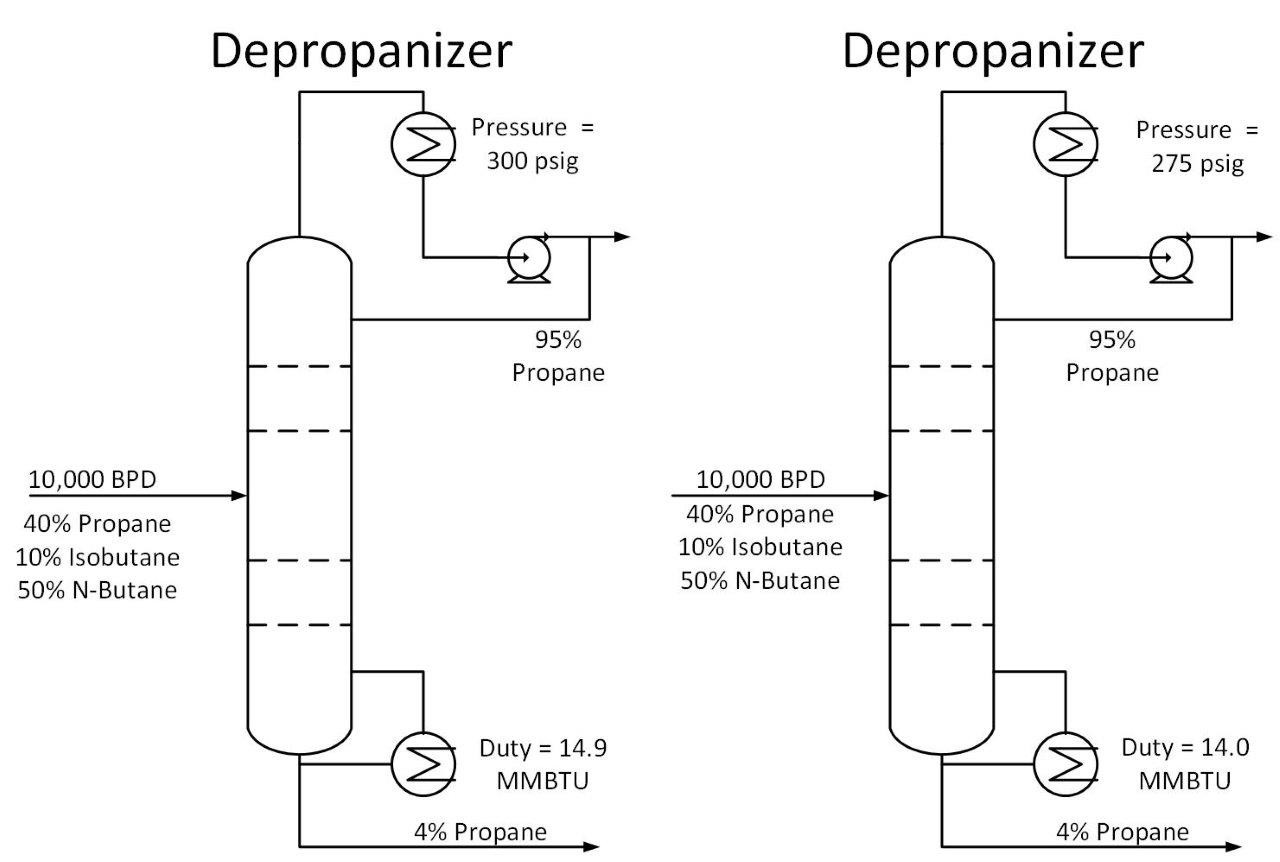 One thing to consider when reducing tower pressure is that the reduction in tower pressure will cause an increase in vapor velocity inside the tower. This increases the chance of flooding inside the tower.
One thing to consider when reducing tower pressure is that the reduction in tower pressure will cause an increase in vapor velocity inside the tower. This increases the chance of flooding inside the tower.